User input¶
BONAFIDE can read four different types of input through its read_input() method.
SMILES strings
XYZ files in XMOL format (
*.xyz)Structured data files (SDF,
*.sdf)Molecule objects (from RDKit)
SMILES strings can be used to input 2-dimensional graph representations of molecules, while XYZ and SD files should be used to represent 3-dimensional molecular structures. SD files provide the opportunity to add information on the connectivity of the atoms within the molecule, which is read and processed by BONAFIDE. For maximum flexibility, also RDKit molecule objects can be directly read in - either with 2D or 3D molecular information (that is, with or without conformers). RDKit molecule objects with only 2D coordinate information (2D conformers) are not supported.
XYZ and SD files as well as RDKit molecule objects can contain multiple conformers (of the same molecule) in a single file/object, and BONAFIDE will process the entire ensemble.
Reading a SMILES string¶
>>> from bonafide import AtomBondFeaturizer
>>> f = AtomBondFeaturizer()
>>> f.read_input("O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl", "diclofenac", output_directory="results/diclo_output")
Reading a file¶
For reading an XYZ or SD file, the input_format parameter must be set to “file”. Multiple
possible conformers are automatically handled during processing.
It is also possible to read the (precomputed) energy of the individual conformers for
calculating Boltzmann-weighted and related features. This is possible by specifying the energy (in
kcal/mol, kJ/mol, or Hartree) in the second line of each conformer block (in case of an XYZ file) or
by specifying a property denoted energy (in case of an SD file). By default, the read_energy
parameter is set to False and must be set to True to read the energy data from the file.
Energy data must always be specified as strings containing the value and the respective unit
separated by a space, for example, "-10.5 kcal/mol" or "-1254.21548 Eh".
It is also possible to prune the conformer ensemble based on relative energies by setting the
prune_by_energy parameter.
>>> from bonafide import AtomBondFeaturizer
>>> f = AtomBondFeaturizer()
>>> f.read_input("diclo.xyz", "diclofenac", input_format="file", read_energy=True)
Alternatively, it is possible to add conformer energies with the attach_energy() method after reading the input file (see
Energy and electronic structure data).
Reading an RDKit molecule object¶
For reading an RDKit molecule object, the input_format parameter must be set to “mol_object”.
Only one molecule object can be processed, and multiple conformers of a molecule should be
associated with the same molecule object. As for files, it is possible to read-in conformer
energies by setting the read_energy parameter to True. In this case, the energy data must
be specified as a property (denoted energy) of the respective conformer. The same energy data
formatting requirements apply as for files (see above).
>>> from rdkit import Chem
>>> from rdkit.Chem import AllChem
>>> from bonafide import AtomBondFeaturizer
>>> # Define example mol object with 5 conformers
>>> mol = Chem.MolFromSmiles("O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl")
>>> mol = Chem.AddHs(mol)
>>> AllChem.EmbedMultipleConfs(mol, numConfs=5)
>>> # Execute BONAFIDE
>>> f = AtomBondFeaturizer()
>>> f.read_input(mol, "diclofenac", input_format="mol_object")
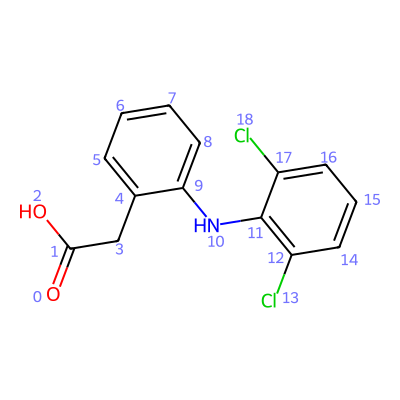
After input reading¶
After a SMILES or file input was read, it is possible to visualize the molecule through the
show_molecule() method. This will
render the molecule in a Jupyter notebook, by default with the indices of the atoms displayed. This
can be changed by setting index_type="bond" (for bond indices) or index_type=None (for no
indices). For 3D structures, it is also possible to look at the molecule and the entire conformer
ensemble, respectively, in an interactive 3D viewer by setting in_3D=True.
>>> from bonafide import AtomBondFeaturizer
>>> f = AtomBondFeaturizer()
>>> f.read_input("O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl", "diclofenac")
>>> f.show_molecule()
It is also possible to inspect the mol_vault attribute of the featurizer object (see the
MolVault class for further details). It collects
all relevant information about the molecule (and its potential conformers).
>>> from bonafide import AtomBondFeaturizer
>>> f = AtomBondFeaturizer()
>>> f.read_input("O=C(O)Cc1ccccc1Nc1c(Cl)cccc1Cl", "diclofenac")
>>> f.mol_vault
...